近日,复旦大学药学院王永辉研究员课题组与华东师范大学生命科学学院卢伟强副研究员课题组合作,在药物化学领域权威期刊《Journal of Medicinal Chemistry》上发表了题为“Design, Synthesis, and Bioevaluation of 2-Aminopteridin-7(8H)-one Derivatives as Novel Potent Adenosine A2A Receptor Antagonists for Cancer Immunotherapy”的研究论文。该论文发现了一类在高浓度腺苷环境中保持高活性的新型蝶啶酮类A2AR小分子拮抗剂,展现出较好的肿瘤免疫治疗潜力。
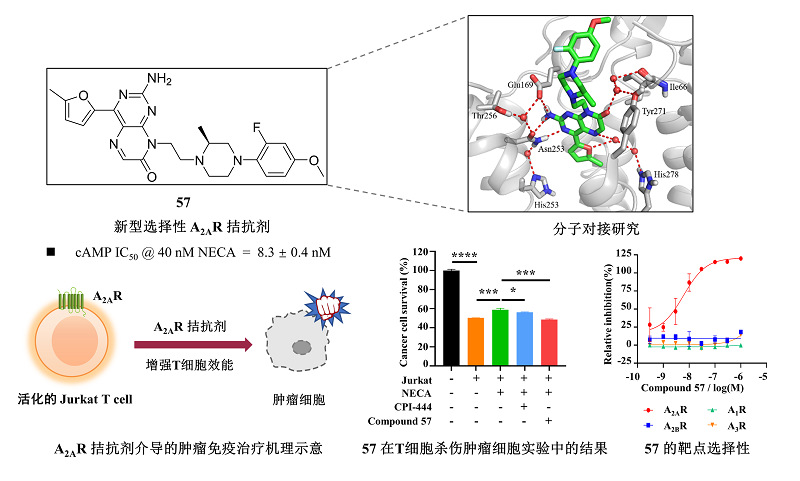
新型蝶啶酮类A2AR小分子拮抗剂化合物57的化学结构、对接模型、活性与选择性及其增强T细胞活化和肿瘤细胞杀伤活性的作用
近年来,腺苷A2A受体(adenosine A2A receptor, A2AR)的肿瘤免疫学调节功能受到广泛关注,A2AR成为极具潜力的肿瘤免疫治疗新兴靶标。A2AR信号通路抑制T细胞的活化和效应功能,削弱抗肿瘤免疫反应,诱导肿瘤免疫逃逸。目前,临床上已有多个A2AR小分子拮抗剂正在开展相关抗肿瘤研究。但是,肿瘤微环境(TME)中的腺苷浓度通常达到微摩尔水平,比正常生理环境中的浓度高出10-100倍,同时也远超帕金森氏症(PD)病理状态下的腺苷浓度,大大限制了A2AR拮抗剂在肿瘤免疫治疗中的效力。因此,开发能够在高浓度腺苷环境中保持高活性的新型A2AR拮抗剂对开发肿瘤免疫治疗具有重要意义。
本研究通过筛选发现具有μM水平A2AR拮抗活性的苗头化合物,采用骨架跃迁、构象限制等药物设计策略进行多轮改造,发现具有nM水平A2AR拮抗活性的先导化合物57(IC50 = 8.3 nM),比临床II期化合物CPI-444活性强10倍(IC50 = 86.5 nM)。化合物57是A2AR的高选择性拮抗剂,对A1R,A2BR,A3R的选择性均超过1000倍。值得注意的是,57能够在模拟肿瘤微环境的高浓度腺苷条件下,即1 μM NECA(5'-N-乙基甲酰氨基腺苷,一种强效A2AR激动剂)刺激下,仍保持对A2AR较好的拮抗活性(IC50 = 179.6 nM)。
进一步研究发现,化合物57能够显著增强T细胞的活化和肿瘤细胞杀伤活性。IL-2作为一种重要的T细胞功能调控因子,在A2AR被NECA激活后,表达量明显下降。化合物57通过拮抗A2AR能够明显恢复IL-2的表达量。在肿瘤细胞和T细胞共培养的实验中,化合物57也能明显逆转NECA对T细胞杀伤肿瘤的抑制作用,从而增强T细胞免疫。综上,化合物57展现出了一定的肿瘤免疫治疗潜力,为针对A2AR的肿瘤免疫疗法的发展提供了新思路。
复旦大学药学院药物化学系2017级博士生余发志、2019级硕士生祝晨宇,华东师范大学生命科学学院2020级硕士生迮书茵为本文的共同第一作者;复旦大学药学院王永辉研究员、谢琼副教授和华东师范大学卢伟强副研究员为本文的共同通讯作者。相关研究工作得到了国家自然科学基金、上海市自然科学基金、国家科技重大专项等的资助。
原文链接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c02199
王永辉研究员课题组介绍
复旦大学药学院药物化学系王永辉研究员课题组主要研究方向是自身免疫病与肿瘤免疫治疗小分子药物的发现及有机与药物化学新方法新技术的研究。现有校特聘研究员1名,副教授2名,在读博士研究生4名、在读硕士究生4名,已毕业博士5名、硕士4名。课题组先后承担国家科技重大专项、国家自然科学基金面上项目、上海市科委生物医药领域科技支撑项目、上海市自然科学基金等科研项目20余项,累计经费超过1100万元。近5年在J Med Chem, Eur J Med Chem, J Org Chem, ACS Med Chem Lett等学术期刊发表SCI论文20余篇;申请中国专利13项(已获5项授权),2项国际PCT专利申请进入美国、日本、欧洲(已获5项国际专利授权)。在自身免疫病和肿瘤免疫小分子药物研究中,发现多个具有开发潜力的候选药物,正在进行临床前评价。

