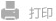Covalent inhibitors have emerged as a promising therapeutic strategy due to their ability to form covalent bonds with target proteins. These inhibitors offer several advantages, including prolonged efficacy, precise targeting, and the potential to overcome drug resistance. However, the inherent reactivity of covalent compounds, while contributing to these benefits, also presents challenges such as off-target effects and potential toxicity. Accurately predicting and modulating this reactivity has become a central challenge in the field.
Recently, the team led by Renxiao Wang, alongside Yan Li and Yifei Qi, from the School of Pharmacy at Fudan University, made significant strides in predicting the reactivity of covalent compounds. By employing deep data mining and machine learning techniques, the team developed an efficient predictive model, named FP-Stack, which serves as a powerful tool for optimizing covalent drugs. Their findings have been published in the Journal of Chemical Information and Modeling.
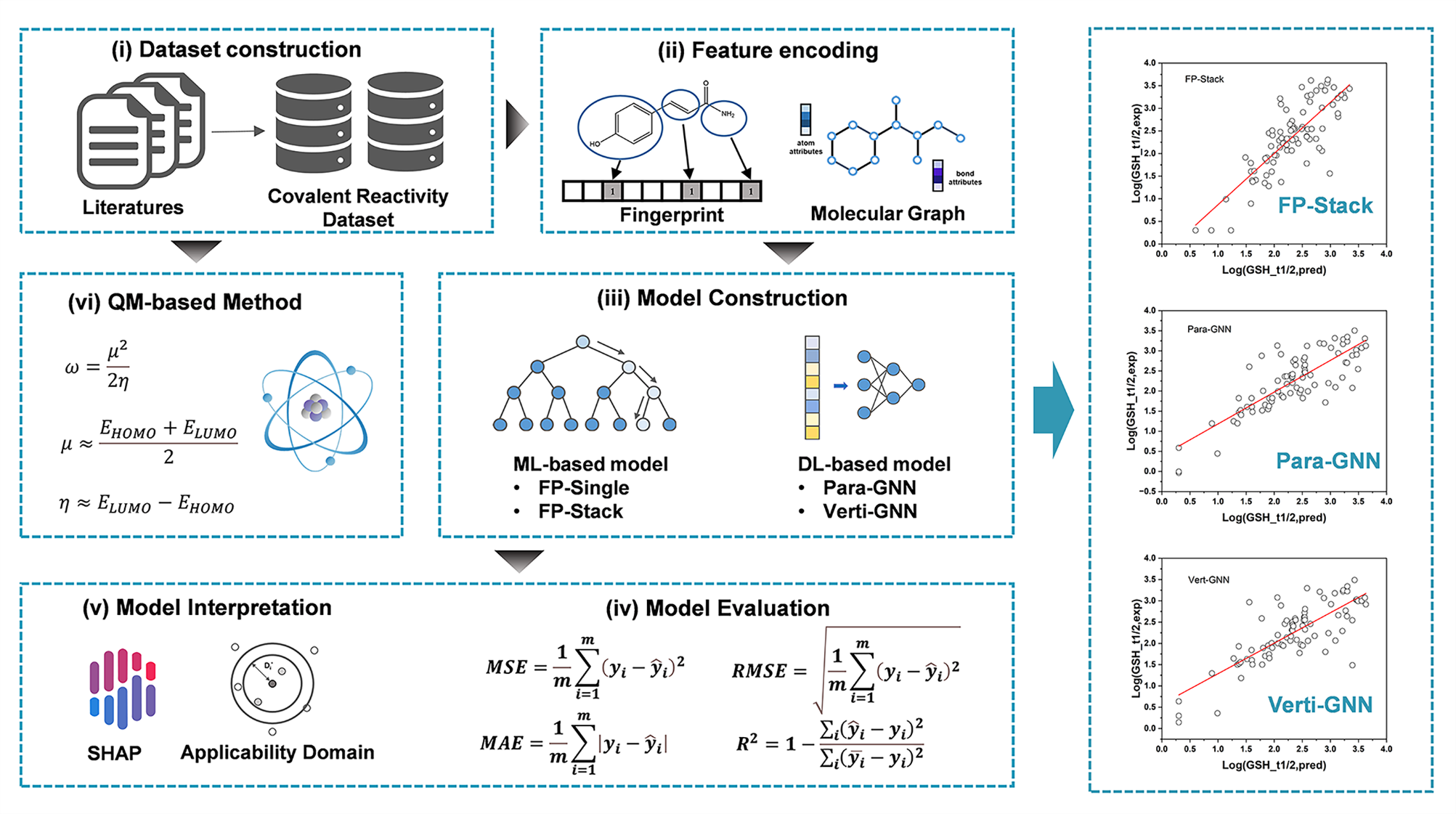
Figure 1 Schematic diagram of the research workflow and the prediction results of three representative models
The team began by systematically reviewing literature related to the reactivity of covalent compounds, sourced from the Web of Science database. After filtering the data through automated scripts and manual review, they curated a high-quality dataset consisting of 419 cysteine-targeting covalent compounds. Based on this dataset, they selected seven molecular fingerprints and seven machine learning algorithms to establish multiple single machine learning models. They then applied an ensemble learning algorithm, using the best-performing individual models as base learners, with a linear regression model as the meta-model, to build the FP-Stack model. Additionally, the team developed four deep learning models, including Graph Convolutional Networks (GCN), Graph Isomorphism Networks (GIN), Graph Attention Networks (GAT), and GT (using the TransformerConv layer from Pytorch Geometric), and explored both vertical and parallel architectural designs. Comparative validation demonstrated that the FP-Stack model outperformed the other models in predictive accuracy and demonstrated exceptional domain extrapolation capabilities.
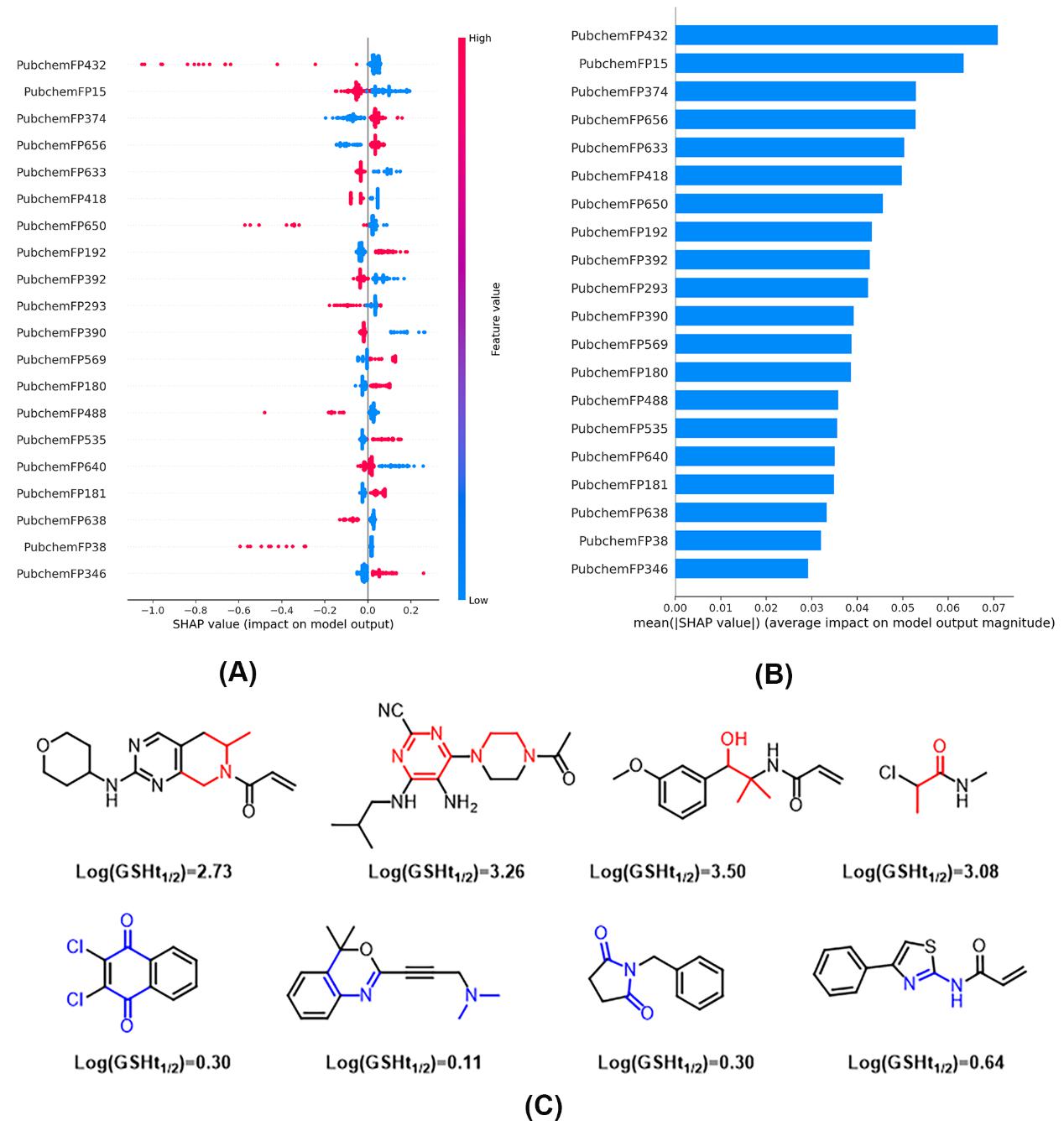
Figure 2. Results of the SHAP analysis of the model. The top 20 SHAP value distribution (A) and average (B) of all samples in the dataset, as well as compounds with representative substructure features (C). Red and blue features indicate weakening and strengthening of the compound's reactivity, respectively.
Further analysis of the FP-Stack model using the SHAP method identified the top 20 molecular descriptors and their associated substructural features that most influenced the model’s predictions. These insights offer valuable guidance for the optimization of covalent drugs. To validate the model's practical utility, the team experimentally tested the glutathione (GSH) reaction half-lives of 10 novel covalent compounds predicted by FP-Stack. The error metrics (MAE, MSE, and RMSE) between experimental and predicted values were close to the performance levels of the best models, confirming the model's robust predictive capabilities.
The first author of the paper is Zhe Zhang, a 2021 master's student from the Department of Medicinal Chemistry at Fudan University. Corresponding authors include Research Associate Professor Yan Li, Researche Professor Renxiao Wang, and Research Associate Professor Yifei Qi, all from the School of Pharmacy at Fudan University. This research was supported by the National Key R&D Program of China, the National Natural Science Foundation of China, and the Shanghai Natural Science Foundation.
Original link: https://pubs.acs.org/doi/10.1021/acs.jcim.4c01591?articleRef=control