随着人工智能的快速发展,深度神经网络能够从蛋白质结构或序列数据中学习高阶特征,深入掌握蛋白质序列与结构的关系,从而实现功能蛋白的高效设计。然而,大多数深度学习模型仍主要用于单链骨架的设计,难以充分应对更复杂的蛋白-蛋白相互作用(protein-protein interaction, PPI)设计挑战。为此,复旦大学药学院戚逸飞/王任小团队开发了专门用于PPI界面序列设计的模型——ProBID-Net。该模型能够根据给定的受体蛋白结构,预测配体蛋白界面上的氨基酸序列,并在多项测试中展现了优良的性能。近日,该项研究成果已在线发表于Chemical Science期刊。

ProBID-Net首先提取目标残基及其周围相邻链残基主链原子在三维网格中的密度分布,随后利用20种天然氨基酸的原子密度信息编码,作为界面相互作用特征的输入,最终输出各位置氨基酸的概率,从而实现界面序列的精确设计。
界面序列平均恢复率和困惑度用于评估序列设计模型对PPI界面氨基酸残基的识别和设计能力。测试结果显示,ProBID-Net和2024年诺贝尔化学奖得主David Baker教授研究小组开发的ProteinMPNN在三个测试集上的界面残基恢复率相当,但ProBID-Net具有更低的困惑度,预测置信度更高。
除此之外,ProBID-Net在零样本训练的情况下,具备预测蛋白复合物结合亲和力变化的能力,并且设计的蛋白序列在界面疏水位点的保守性更高,且更符合天然蛋白中氨基酸的分布和替换规律。经过AlphaFold2 multimer验证,ProBID-Net设计的序列能够良好地折叠为原复合物结构,且链间PAE较低,进一步表明ProBID-Net在结合界面设计方面优于ProteinMPNN (图1)。该模型为蛋白-蛋白相互作用的设计提供了一个有效工具。目前,ProBID-Net已开源,地址为:https://github.com/ComputArtCMCG/ProBID-NET。
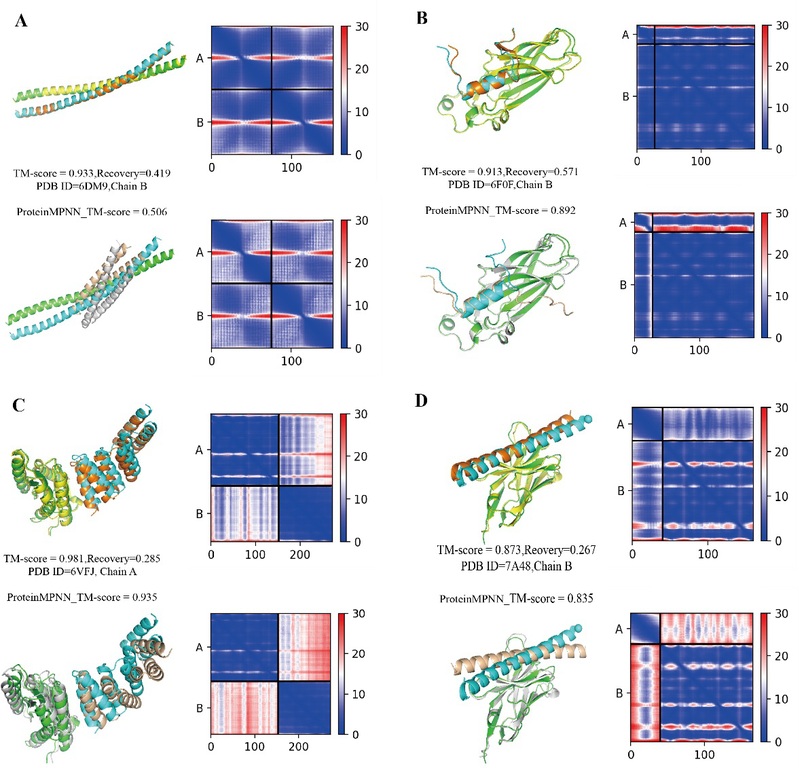
ProBID-Net(上)和ProteinMPNN(下)设计序列预测结构叠合比对与AlphaFold-Multimer预测链间PAE热图
复旦大学药学院药物化学系2021级硕士研究生陈志航为本论文第一作者,复旦大学药学院王任小研究员、戚逸飞副研究员为本论文的通讯作者。该研究工作得到了国家自然科学基金委、国家重点研究计划、上海市科委的项目资助,并采用复旦大学CFFF平台进行计算。
原文链接:ZH Chen, et al. ProBID-Net: A Deep Learning Model for Protein-Protein Binding Interface Design. Chemical Science, 2024.
https://pubs.rsc.org/en/Content/ArticleLanding/2024/SC/D4SC02233E
